| Introduction | Jiri Klimes | Research | Members | Theses available | Teaching | Publications |
I'm always available to discuss possible projects and Bc/MSc/PhD theses themes. There are several options available, listed below, which include calculations, code development, and analytic derivations. If you are interested or you would just like to know more, let me know.
Available themes (any level)
- Ab-initio description of molecular solids
- Accurate reference data for solid state chemistry
- Weak interactions in density functional theory
- Asymptotic corrections for quantum chemical methods
SFG Projects
|
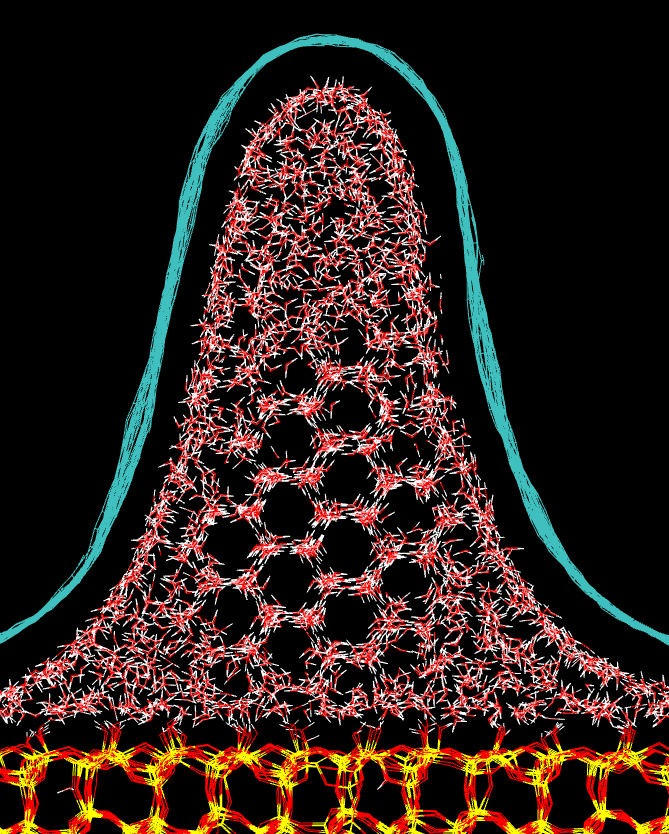
We all know that bulk water freezes at 0 degree Celsius. However, when we confine water in a tube or tunnel with a diameter of few nanometres its freezing temperature can drop by tens of degrees. Moreover, the freezing temperature can differ in different parts of the tube or tunnel, such as in the graphene wrinkle shown in the picture. There are several topics that can be studied for such systems. For example, we can use computer simulations to understand how the flexibility and surface of the tube or tunnel affect the freezing temperature. As it's very difficult to freeze water in computer simulations, even below zero Celsius, another topic could be to to test if the freezing can be induced by fixing some of the water molecules in the structure of ice. |
|
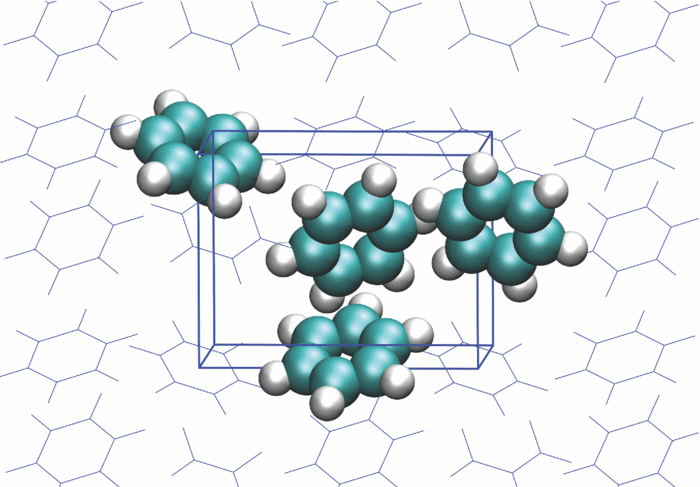
Simulations of complex systems are often performed using simplified models of molecules capturing electrostatic interactions, attractive dispersion forces, and repulsive short range interactions. While they are computationally cheap, the models are often too simple to describe the fine details of the interactions. Recent years have seen a growing interest into models based on neural networks or other machine learned potentials. They offer higher accuracy at computational cost lower than high-level methods based on quantum mechanics. The goal of this project will be to test some of these approaches for modelling interactions in molecular solids. |
|
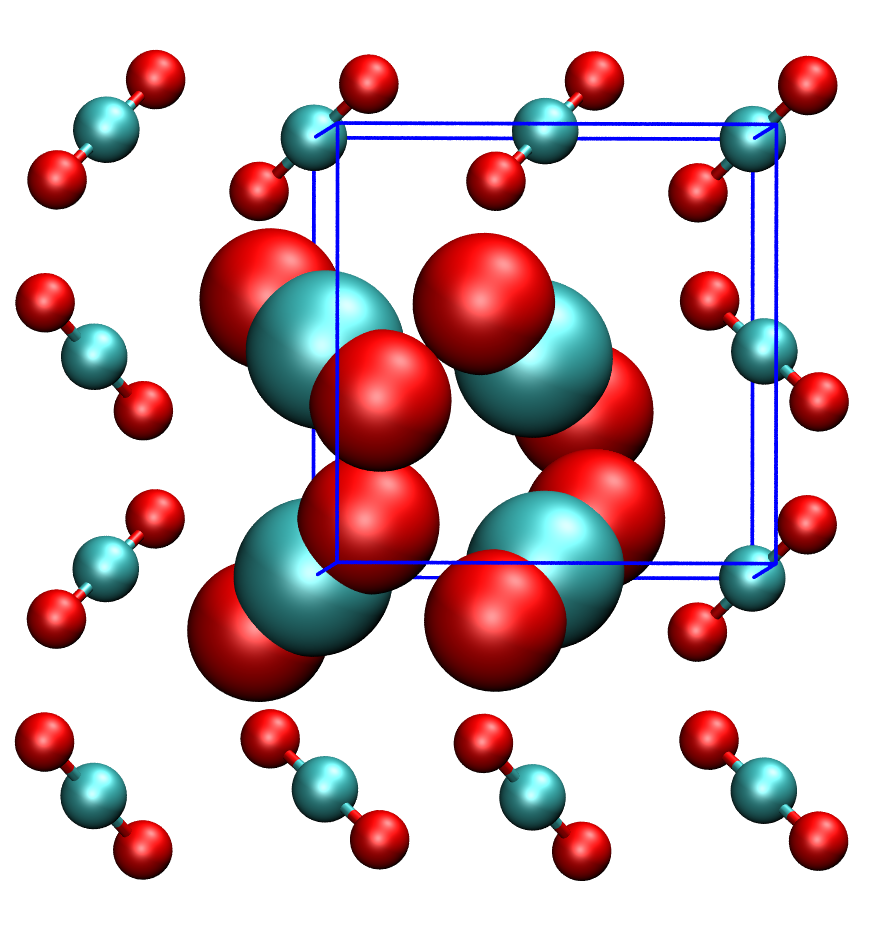
Carbon dioxide is a very important molecule. It is therefore necessary to be able to accurately describe its interactions with different materials. The most widely used methods for computer simulations of materials surprisingly fail in accurately describing the interactions even in the simplest system involving carbon dioxide, its crystal phase (the dry ice). In this project we will analyze the interactions between the molecules in the CO2 crystal to find out what is the cause of this problem. |
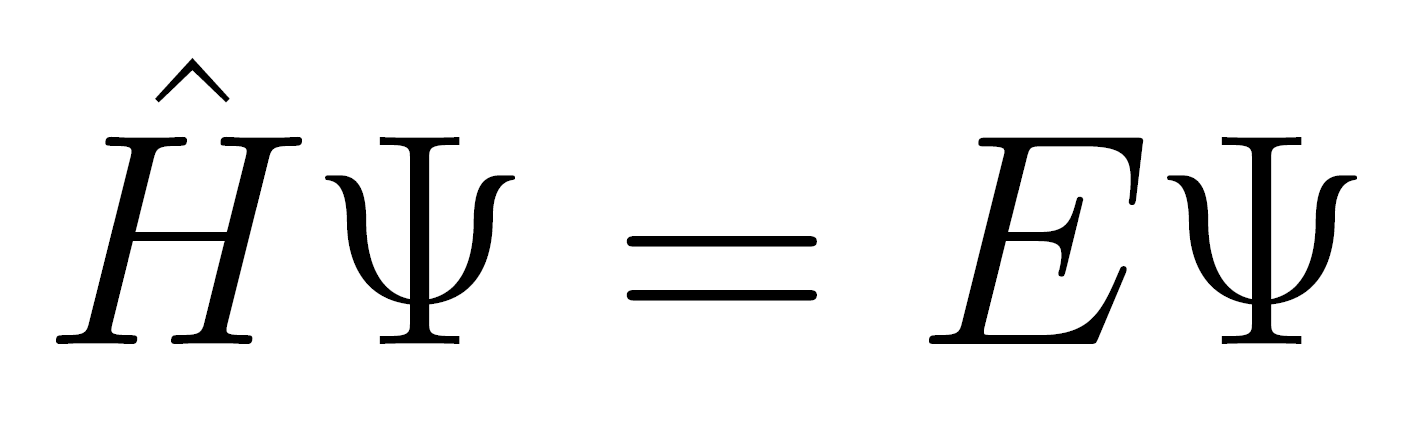
|
To goal of this project is to create interactive Jupyter notebooks with homeworks given in the lecture course Introduction to quantum mechanics taught in the summer term of the second year of BSc. Some of the homeworks are on this page. Examples of such notebooks can be found for example here (files ending with ipynb) or here for a bit advanced topics. |
PhD theses
|
|
One often finds differences when comparing published binding energies of molecular solids. And the differences can be large, in fact, terribly large. They can be even found for results published with the same computational package! One of the goals of this project is to understand the limits of precision of widely used programmes and identify settings that allow trustworthy results to be obtained. This will allow the candidate to make reliable predictions of important properties of molecular solids, such as of the transition pressures between high pressure phases of matter. |
- Understanding the accuracy of quantum chemical methods for the description of adsorption (funded via Primus)
- Done by Kyrylo Prokofiev (2017-
The goal of this project is to understand the strenghts and weaknesses of different quantum chemical methods for the description of adsorption on surfaces. Currently, density functional theory (DFT) is the most widely used method for studies of molecular adsorption on surfaces of solids. However, even the most recent methods can give adsorption structures or energies that are qualitatively incorrect and their predictions should be checked for every specific system. More advanced methods, such as the second order perturbation theory (MP2) or the random phase approximation (RPA) should improve over DFT in terms of accuracy. However, how much do they improve and what are their limits is unknown.
- QM/QM description of adsorption in porous materials (funded via Primus)
The goal of this project is to develop schemes that would allow one to obtain highly accurate adsorption energies of molecules in extended porous systems. Other properties, such as vibrational or NMR spectra will be also studied. The project will explore combinations of quantum chemical methods in a QM/QM setting, reliying on quantum chemical methods that have been recently implemented within periodic boundary conditions.
MSc theses
- Convergence of the embedding scheme
- Done by Jaroslav Hofierka (2017-2019)
- Accurate binding energies of molecular solids
- Asymptotic convergence of basis set expansion of quantum mechanical methods
BSc theses
- Cluster expansion versus periodic calculations of binding energies of molecular solids
- Done by Jaroslav Hofierka (2016-2017)
- Available under "Text prace" here